Chromosome painting using non admixed ancestral accessions (VCFHunter)
The aims of this tutorial are to showing how data should be processed to be then visualized with the GeMo
Installation
Install VCFHunter following the documentation presented above:
git clone https://github.com/gdroc/GeMo_tutorials.git
cd GeMo_tutorials
python3 -m venv $PWD/venv
source venv/bin/activate
pip install numpy
pip install matplotlib
pip install scipy
Download datasets
Two ways :
Download Baurens_et_al_2019.zip available on Zenodo
mkdir data
cd data
wget https://zenodo.org/record/6542870/files/Baurens_et_al_2019.zip
unzip Baurens_et_al_2019.zip
ls Baurens_et_al_2019.vcf > Vcf.conf
Goto Identification of private alleles and formatting output for more analysis
Create input dataset using Gigwa, a web application for managing and exploring high-density genotyping data, to download a VCF
Select the database Populations_A_B
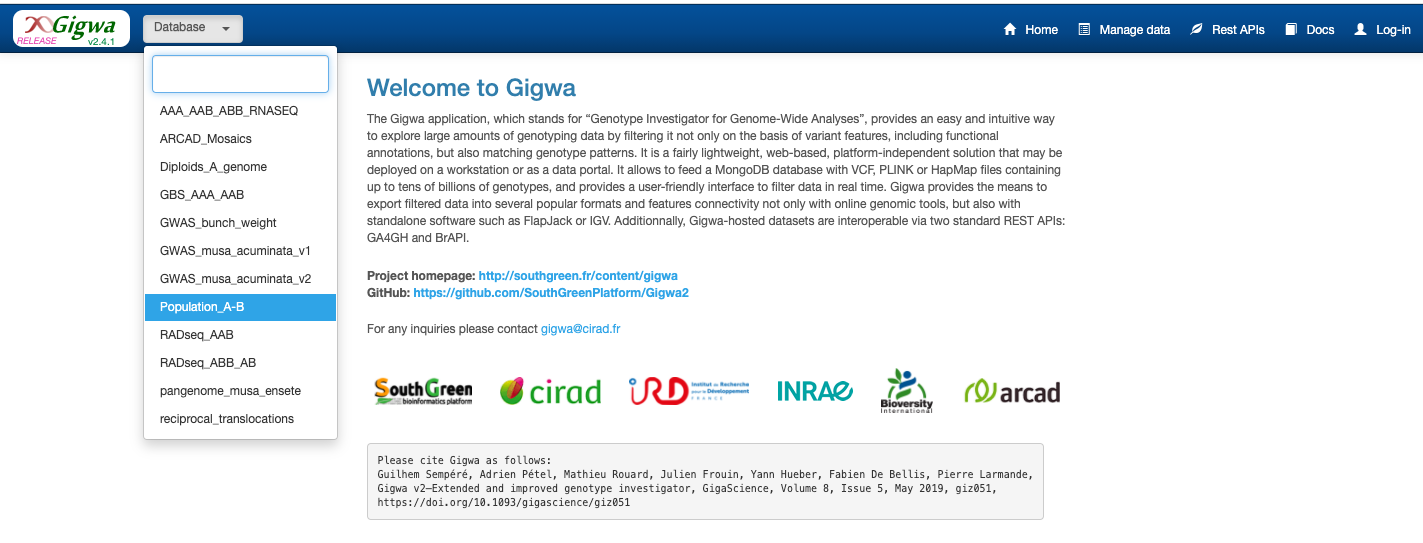
Select the accessions P2 and T01 to T11 on the Indivuals drop down menu, and click on Search button
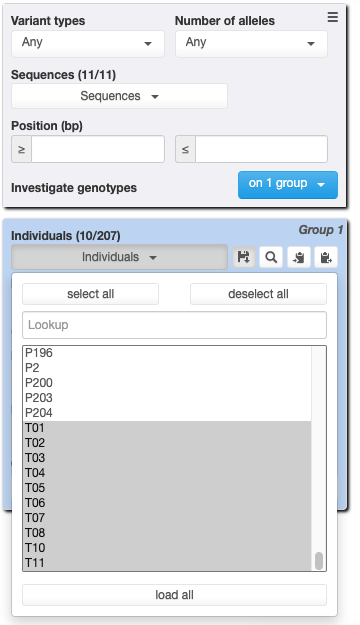
Download result (check radio “Export Metadata” and “Keep file on servers”)
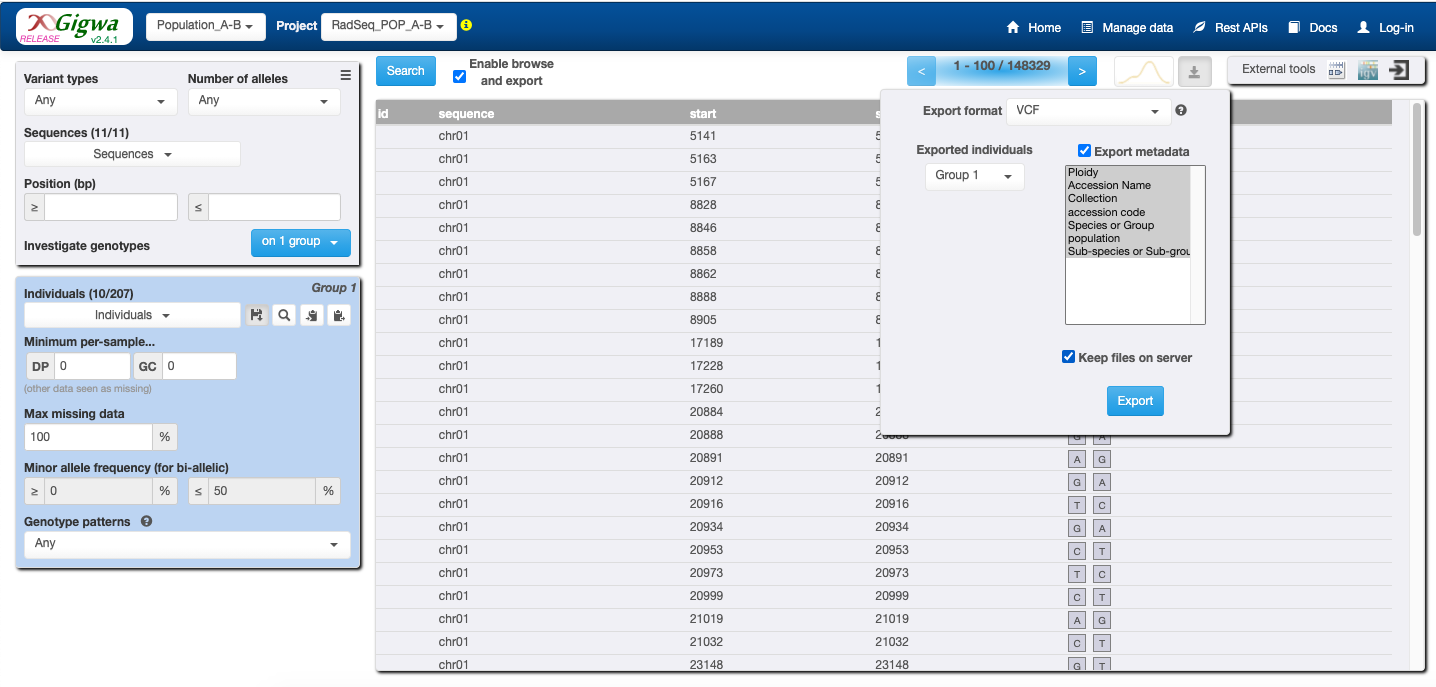
Copy the link, and create a repository on your terminal
mkdir data
cd data
wget --no-check-certificate https://www.crop-diversity.org/gigwa/genofilt/tmpOutput/anonymousUser/b429763f507dc1bb2b169d7da5cf1804/Population_A-B__project1__2021-10-12__148329variants__VCF.zip
unzip Population_A-B__project1__2021-10-12__148329variants__VCF.zip
cut -f 1,6 Population_A-B__21individuals_metadata.tsv > Baurens_et_al_2019_origin.txt
sed -i 's:balbisiana:BB:' Baurens_et_al_2019_origin.txt
sed -i 's:acuminata:AA:' Baurens_et_al_2019_origin.txt
ls Population_A-B__148329variants__10individuals.vcf > Vcf.conf
VCF content
grep "^#CHROM" Population_A-B__148329variants__21individuals.vcf
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT ACC48-FPG ACC48-FPN ACC48-P_Ceylan ACC48-Red_Yade DYN163-Kunnan DYN275-Pelipita DYN359-Safet_Velchi GP1 GP2 P1 P2 T01 T02 T03 T04 T05 T06 T07 T08 T10 T11
Workflow
The principle of this analysis is to :
Identify specific allele of distinct genetic pools,
Calculate the expected allelic ratio of these alleles in these genetic pools,
Calculate the observed allelic ratio a/several given accessions
Normalize these observed ratios using expected ratio to infer the number of haplotypes of each genetic pools that are present on a given windows of the studied accession.
Files obtained at the end of the process can be given to GeMo tools to visualize data and optimize parameters.
Input
Baurens_et_al_2019_origin.txt
Vcf.conf is a file which contained path to vcf files which will be used for e-chromosome painting.
Baurens_et_al_2019_chromosome.txt (tabulated file with the chromosome name and length)
Baurens_et_al_2019_color.txt
group |
name |
r |
g |
b |
|---|---|---|---|---|
AA |
acuminata |
0 |
255 |
0 |
BB |
balbisiana |
255 |
0 |
0 |
Identification of private alleles and formatting output for more analysis
bin/IdentPrivateAllele.py -c data/Vcf.conf -g Baurens_et_al_2019_origin.txt -o step1 -a y -m y
In this first step, the program use genotyping information provided in vcf files passed in Vcf.conf file and the file Origin.tab containing the corresponding genetic pools of some accessions of the vcf to identify alleles specific of each pools.
Outputs can be found in directory passed in -o option. For each accessions identified as belonging to a genetic pool a directory is created.
tree step1
step1
├── P2
│ ├── P2_ratio.tab.gz
│ └── tmp_1_P2_stats.tab
├── T01
│ ├── T01_ratio.tab.gz
│ └── tmp_1_T01_stats.tab
├── T02
│ ├── T02_ratio.tab.gz
│ └── tmp_1_T02_stats.tab
├── T03
│ ├── T03_ratio.tab.gz
│ └── tmp_1_T03_stats.tab
├── T04
│ ├── T04_ratio.tab.gz
│ └── tmp_1_T04_stats.tab
├── T05
│ ├── T05_ratio.tab.gz
│ └── tmp_1_T05_stats.tab
├── T06
│ ├── T06_ratio.tab.gz
│ └── tmp_1_T06_stats.tab
├── T07
│ ├── T07_ratio.tab.gz
│ └── tmp_1_T07_stats.tab
├── T08
│ ├── T08_ratio.tab.gz
│ └── tmp_1_T08_stats.tab
├── T10
│ ├── T10_ratio.tab.gz
│ └── tmp_1_T10_stats.tab
└── T11
├── T11_ratio.tab.gz
└── tmp_1_T11_stats.tab
Determination of expected read ratio for each ancestral position based on ancestral accessions merged together
bin/allele_ratio_group.py -g Baurens_et_al_2019_origin.txt -p _ratio.tab.gz -o step2 -i step1
In this second step the program take the input of specific allele identified in each accessions used to define genetic pools (ratio.tab.gz files of step1 folder) and calculate an average expected allele ratio (globally a proxy of the fixation level of the allele) in the genetic pool the allele belongs.
A tabulated file is generated per genetic pool with the following format:
c hromosome |
position |
allele |
genetic pool |
average allelic ratio observed |
number of ancestral a ccessions |
|---|---|---|---|---|---|
chr02 |
15033812 |
A |
AA |
0.9959677 419354839 |
8 |
chr02 |
17722345 |
G |
AA |
1.0 |
8 |
chr09 |
39501254 |
T |
AA |
1.0 |
8 |
chr05 |
17536961 |
T |
AA |
1.0 |
8 |
chr06 |
10144735 |
A |
AA |
0.9931737 588652483 |
8 |
chr08 |
4718673 |
T |
AA |
0.9932432 432432432 |
8 |
chr10 |
37498708 |
T |
AA |
0.9239074 518611573 |
8 |
Calculation of observed ratio in other accessions
The third step is to calculate, for each position in which an allele specific of a genetic pool was identified, the observed allelic ratio in a studied accession. In this example we calculate this ratio on the Kunnan accession.
bin/allele_ratio_per_acc.py -c Vcf.conf -g Baurens_et_al_2019_origin.txt -i step2 -o step3 -a DYN163-Kunnan
The output can be found in the step3 folder passed in -o option. This tabulated file contained 6 columns: column 1 corresponded to the chromosome, column 2 is the position of the allele, column 3 is the allele, column 4 corresponded to the observed allele frequency in the accession, column 5 is the expected allele frequency calculated at step 2 and column 6 is the genetic group to which the allele has been attributed.
For example : zmore step3/DYN163-Kunnan_ratio.tab.gz
chr |
pos |
allele |
obs_ratio |
exp_ratio |
grp |
|---|---|---|---|---|---|
chr01 |
20888 |
A |
0.0 |
0.23513227513227516 |
BB |
chr01 |
20916 |
C |
0.14754098360655737 |
0.28604868303910713 |
BB |
chr01 |
21019 |
G |
0.21875 |
0.3700537473602161 |
BB |
chr01 |
67413 |
T |
0.5818181818181818 |
1.0 |
AA |
chr01 |
67413 |
A |
0.41818181818181815 |
1.0 |
BB |
chr01 |
67461 |
G |
0.0 |
0.975 |
AA |
chr01 |
89923 |
G |
0.6842105263157895 |
1.0 |
AA |
chr01 |
89923 |
T |
0.3157894736842105 |
1.0 |
BB |
chr01 |
89958 |
T |
0.6842105263157895 |
1.0 |
AA |
Calculation on sliding of the normalized observed ratio and ancestral blocs
In this step, in a given sliding windows, the observed average allelic ratio is calculated for each genetic pool and normalized by the expected allelic ratio. The resulting value is used to infer the number of haplotypes from the studied genetic pool present in the studied accession.
Output are of two types:
<accession>_win_ratio.tab.gz file containing normalized values for each genetic pools in the given windows. This file contained 4 + X columns, X being the number of genetic pools tested. The column 1 contained the chromosome name, column 2 contained the position of the central allele in the windows, column 3 contained the start position of the windows and column 4 contained the end position of the windows. Columns 5 to end contained the normalized ratio calculated for the accessions. A columns per genetic pool.
<accession>_<chromosome>_<haplotype>.tab contained the hypothesized haplotypes from this accession given results from tab.gz file. Haplotype are hypothetic ones that tries to minimize recombinations events between distinct genetic pools. These files are formatted as follows: column 1 contained accession name, column 2 contained chromosome ID, column 3, 4 and 5 contained start, end, and origin of a region.
mkdir step4
bin/PaintArp.py -a DYN163-Kunnan -r step3/DYN163-Kunnan_ratio.tab.gz -c Baurens_et_al_2019_color.txt -o step4/DYN163-Kunnan -w 12 -O 0 -s Baurens_et_al_2019_chromosome.txt
File formatting for GeMo visualization
This steps aims at reformatting the files so that they are compatible with GeMo tool. GeMo tool performs two tasks, the first one consists in drawing ancestral block identified at step 4. The second one also draw these blocks but allowed refinement of these block using custom and adjustable parameters. For block drawing of step 4 we will reformat block files so that they match expectation with GeMo. For this run the following command line:
mkdir step5
bin/convertForIdeo.py --name DYN163-Kunnan --dir step4 --col Baurens_et_al_2019_color.txt --size Baurens_et_al_2019_chromosome.txt --prefix step5/DYN163-Kunnan --plo 2
This command generate several files with the following names:
<accession_id>_ideo.txt that contained block that could be drawn with GeMo (data section),
<accession_id>_curve.txt that contained block that could be drawn with GeMo (data section),
<accession_id>_ideoProb.txt that contained block that could be drawn with GeMo (data section),
<accession_id>_chrom.txt that contained information required to draw chromosomes.
<accession_id>_color.txt contained color information that could be used to draw blocks with custom color.